Poster (P71)
Mechanistic Study on Non-Oxidative Methane Dehydrogenation and C-C Coupling for Ethylene Formation
Sonit Balyan 1, Sourabh Mishra 1, Tuhin S. Khan1, M. Ali Haider1, K. K. Pant1
1Department of Chemical Engineering, IIT Delhi, India
DFT calculation have been employed to study the CH4 activation and C-C coupling for ethylene formation on Mo4C2 cluster. Methane is activated on the Mo4C2 cluster with an intrinsic activation barrier ~ 116 kJ/mol, related to the second methane activation. The highest intrinsic barrier was obtained for the C-C coupling step forming C2H6 (~ 151 kJ/mol). The activation of C2H6 (~70 & 95 kJ/mol) was relatively easy and C2H4 will be formed easily on the catalyst surface. All the energy barriers observed for these elementary steps have been shown in Figure 1. Also C-H bond activation barrier decreases as the Mo4C2 cluster is more reduced in nature (less positive). A detailed mechanistic understanding is developed that correlates the effect of charge on catalyst activity. It dictates that lesser residual charge on MoxCy cluster lowers the barrier for methane dehydrogenation.
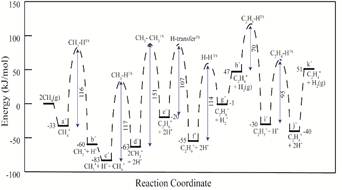
Figure 1. Reaction diagram for methane dehydrogenation and C-C coupling to form ethylene on Mo4C2 cluster